Introducción
El síndrome de microdeleción 16p11.2 proximal es una anomalía cromosómica relacionada con TEA caracterizada por un retraso en el desarrollo y el lenguaje, niveles variables de discapacidad intelectual, rasgos dismórficos leves y predisposición a la obesidad1,2. Este síndrome presenta dificultades diagnósticas relacionadas con la interpretación de la variación genética y la variabilidad clínica de presentación. Además, su manejo es multidisciplinar.
Caso clínico
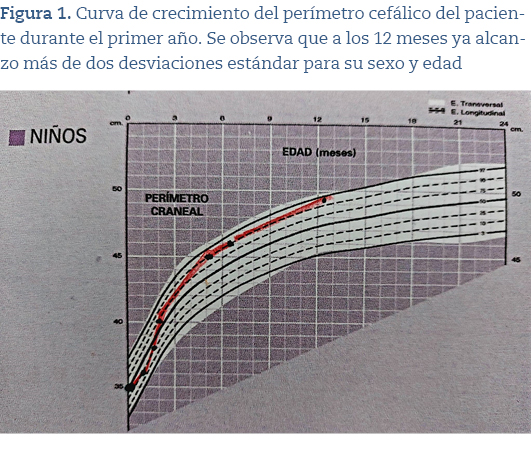
Presentamos a un niño de 7 años, primogénito, de padres sanos no consanguíneos cuya gestación fue controlada, con nacimiento a las 38 semanas por cesárea urgente por sospecha de pérdida de bienestar fetal que no precisó reanimación con antropometría normal al nacimiento. A los 5 meses presentó, durante un día, 5 episodios de crisis tónico-clónicas generalizadas, de 1-3 minutos de duración, que cedieron con la administración de diazepam rectal, con períodos poscríticos de menos de 1 hora sin fiebre. Se mantuvo ingresado en la planta de pediatría durante 4 días con analítica sanguínea, estudio de líquido cefalorraquídeo, electroencefalograma y tomografía computarizada (TC) cerebral sin hallazgos patológicos. Posteriormente, fue dado de alta con diagnóstico de crisis clónicas generalizadas y macrocefalia benigna del infante (perímetro cefálico 45 p90) y se pautó trileptal. Tras ello no volvió a presentar convulsiones y al año se le retiró el tratamiento anticonvulsivante.
Todos los estudios de control posteriores han sido normales, aunque durante el seguimiento en neurología se objetiva hipotonía y retraso global del neurodesarrollo (sedestación a los 9 meses, inicia la marcha a los 19 meses y primeros bisílabos referenciales con 2 años), por lo que se deriva a rehabilitación y en Atención Primaria se indica evaluación en el Centro Regional de Coordinación y Valoración Infantil (CRECOVI), donde recibe apoyo en logoterapia y psicomotricidad hasta ahora. En este momento tiene reconocido un grado de discapacidad del 20% y se encuentra escolarizado en colegio ordinario. No ha precisado adaptación curricular y no se han objetivado alteraciones del comportamiento.
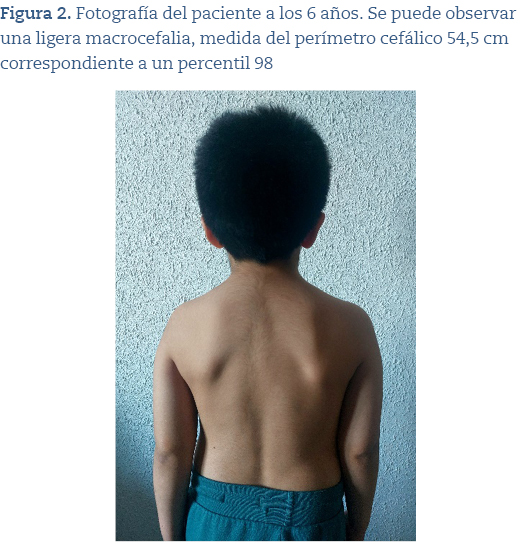
Hace 1 año es derivado a consulta de genética tras objetivar en consultas de neurología y pediatría de Atención Primaria macrocefalia (54,5 cm p98) (figuras 1 y 2) y rasgos faciales sutiles (hendiduras palpebrales cortas, pestañas largas). Se le hizo estudio de hibridación genómica comparada con array de 60.000 oligonucleótidos y se detectó un intervalo de material genómico en la región cromosómica 16p 11.2, de un tamaño aproximado de 524.5 kb, que se encuentra en un número de copias correspondiente a una deleción en mosaico. También se amplió el estudio a sus progenitores con resultado negativo.
Discusión
La microdeleción 16p11.2 es una rara condición genética cuya prevalencia alcanza una prevalencia en la población general del 0,01%3-5. La base de datos europea Orphanet estima una prevalencia de este síndrome en 1/5.000 en la población general y señala que es una etiología común en las personas con TEA, estimada en aproximadamente 1:150 individuos diagnosticados con TEA2. Es una alteración que se expresa de forma heterogénea y que abarca distintos grados del espectro autista, así como retraso del neurodesarrollo, retraso del lenguaje, convulsiones y tendencia a la obesidad. Su manifestación es irregular al tener penetrancia incompleta y expresividad variable3, y es más tenue en personas con mosaicos.
Estas personas pueden presentar una facies característica con macrocefalia, orejas de implantación baja, rostro plano, pliegue transverso, clinodactilia del quinto dedo de las manos, micrognatia o retrognatia, hipertelorismo, cuello corto y sindactilia del segundo y tercer dedo de los pies3. Además, pueden aparecer malformaciones cardíacas, hemivértebras, alteraciones del tracto urinario y digestivas.
Existen dos variantes de la microdeleción del 16p11.2: la típica o más común que afecta aproximadamente a 593 kilobases y la atípica o menos frecuente que es adyacente o distal a la primera y afecta a 22 kilobases.
Esta microdeleción se hereda de forma autosómica dominante, aunque en la mayoría de los casos es una mutación de novo3,6 y las características más frecuentes son retraso del habla y del desarrollo del lenguaje, dificultad del aprendizaje, aumento en la susceptibilidad a alteraciones del espectro autista, rasgos faciales leves, tono muscular débil, tendencia al sobrepeso y ocasionalmente convulsiones7.
La deleción típica se describió en 2007 y afecta a 29 genes. Hasta un 70%, tienen algún rasgo autista y la epilepsia se manifiesta en un 20% de los pacientes8. El diagnóstico está basado en la sospecha clínica y en la evaluación en la unidad de genética mediante estudios genómicos capaces de identificar un número de copias como la hibridación genómica comparativa en microarreglos conocida como array CGH6. El tratamiento es multidisciplinar y conlleva seguimiento por diversas especialidades, así como estimulación precoz, logopedia y manejo nutricional para evitar la obesidad2.
En cuanto al asesoramiento genético, a pesar de que esta deleción mayormente aparece de novo, puede ser heredada de padres afectados, por lo que se debe aconsejar a los padres del caso índice (sobre todo si desean más descendencia)8,9.
El papel del personal médico/pediatría de Atención Primaria es importante como observadores del cuadro clínico, para orientar a la familia y derivarla a los recursos que sean necesarios. Además, la prevalencia basada en reportes internacionales hace pensar que es posible que estemos infradiagnosticando en nuestra población esta entidad, por lo que ha de sospecharse en paciente con déficit intelectual y sobre todo si existe retraso del desarrollo, convulsiones y macrocefalia.
Agradecimientos
Se obtuvo el consentimiento escrito del padre del paciente para la publicación del manuscrito.
